Cyclic Peptide Therapeutics in Drug Discovery
To date, there are over 40 cyclic peptide therapeutics in clinical use, and their number is constantly growing.[1,2] This is because, when peptides undergo cyclisation, they gain unique or enhanced biological and chemical properties. So, in this latest article we explore their unique characteristics and how they differ from their linear counterparts. We also compare them to other drug modalities that are commonly used in drug discovery. Finally, we present an application example using one cyclopeptide that our team recently synthesised for a study the Universities of Birmingham, UK, and Sydney, Australia.
Key differences between cyclic and linear peptides
Although linear and cyclic peptides are formed from similar building blocks, they differ significantly in their properties. The most obvious difference is the increased rigidity of the cyclised structures compared to their linear analogues. The constraint introduced when closing the loop, decreases their conformational flexibility. As a result, this may offer additional benefits compared to their linear counterparts. In particular:
- They display larger surface areas available for interaction with other targets. This is advantageous as it may enhance their binding properties and specificity.
- Cyclisation also increases their resistance to proteolytic degradation. Indeed, backbone cyclisation blocks the peptide’s termini, preventing exopeptidases from hydrolysing it. Additionally, the peptide’s increased rigidity hinders proteolytic enzymes from recognising, binding and cleaving the peptide bonds effectively.
- Peptide cyclisation may also improve their cellular uptake. While most cyclic peptide drugs aim at extracellular targets, some are capable of crossing cell membranes and target intracellular proteins. They are usually small and non-polar, but not always. For example, cyclosporine is an archetype of peptides breaking Lipinski’s rule of 5. Despite its high molecular weight, it is cell permeable and orally bioavailable. The rationale behind this can be explained as follows. Firstly, head-to-tail cyclisation decreases the peptide polarity by removing the two ionisable C and N termini. Secondly, the constrained structures may also adopt conformations favourable to forming intramolecular hydrogen bonds between residues. In return, this may reduce solvation and shield polar surfaces enabling their diffusion across the apolar membrane. This last point is important as membrane permeability is a key factor for oral bioavailability.
Comparisons of cyclic peptide therapeutics with other drug modalities
Cyclisation enables peptides to gain properties that are beneficial in drug discovery. As a result, cyclopeptide drugs occupy a unique space between small molecules and larger biologics such as antibodies. But how do they compare exactly?
- Small molecules are smaller (less than 1kDa) and usually suitable for oral administration. Often hydrophobic in nature, they can pass through the cell membrane and be used for intracellular targets. However, one of the drawbacks is that may lack selectivity especially when targeting only one protein homologue is required.
- On the other hand, biologics such as antibodies have high selectivity. However, due to their large size hindering their diffusion, they are mostly limited to extracellular targets although, conjugation to cell-penetrating peptides may improve transport across membranes.
- Therefore, cyclic peptide drugs are a promising class of molecules that combine many advantages of both large biologics and small molecules. They offer alternatives strategies to target “undruggable” space currently inaccessible by conventional drug modalities. Larger than small molecules, they can bind to flat intracellular protein–protein interactions (PPIs) with antibody-like specificity and affinity. This is key as PPIs control many essential biological pathways. This is exciting as “75% of all disease relevant human proteins including those involved in intracellular protein-protein interactions (PPIs) are undruggable with current drug modalities”.[4,5] But this is not the only reasons why they are gaining popularity. Their relatively ease of synthesis with many Fmoc amino acids and other building blocks commercially available, low cost and low toxicity also contribute to their popularity as attractive drug targets and for other applications.
Case study: Synthesis of a cyclic peptide PPI inhibitor
Recently, our laboratory synthesised peptide OS1 with the amino acid sequence: CTERMALHNLC.[6] It is a short peptide containing terminal cysteine residues, which upon oxidation of the thiol groups, forms a cyclic structure with a disulfide bridge. Originally isolated from a cysteine-constrained phage display library, this peptide is a great PPI inhibitor example. In particular, it inhibits the interaction of platelet glycoprotein Ibα (GPIbα) and the von Willebrand factor (vWF). Although the binding of both proteins is a normal event in haemostasis, conditions such as stroke or myocardial infarction can over activate this pathway, leading to thrombosis. However, targeting the binding site with small molecules is difficult due to the large 2600-Å2 protein-protein interaction. In contrast, the cyclic peptide allosteric inhibitor OS1 was able to disrupt the interaction with sub-nanomolar potency.[7,8]
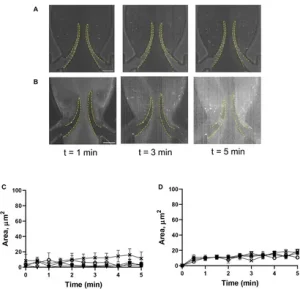
FIGURE – Deposition of platelets is mediated by the VWF-GPIbα axis. Platelets were preincubated with OS-1 peptide (M3456 CTERMALHNLC, AltaBioscience, 12µM) (A), orwith0.1U/ml thrombin+OS-1peptide (B) for 5 min, fluorescently labelled, reconstituted with whole blood, and perfused through a symmetrical valve for 5 min. (C, D), Quantitation of platelet accumulation on more flexible (TL area, circles; TS area, open triangles) and less flexible (TL area, squares; TSarea, Xs) leaflets. Scale bar 100 µm. Error bars represent SEM, n=3.
Unmodified image taken from Hosam Alden Baksamawi, Alessio Alexiadis, Daniele Vigolo and Alexander Brill, “Platelet accumulation in an endothelium-coated elastic vein valve model of deep vein thrombosis is mediated by GPIbα—VWF interaction” Frontiers in Cardiovascular Medicine, vol. 10, 2023, Article 1167884. Under License: Deed – Attribution 4.0 International – Creative Commons
Get in touch
At AltaBioscience, our team has built extensive expertise in custom peptide synthesis, delivering rapid turnaround times and exceptional quality to researchers globally. Operating under ISO-9001 certification, our laboratory follows rigorous quality control measures, guaranteeing consistent reliability for all projects.
For more information about our synthesis or analytical services or to discuss your specific needs, please reach out to us or email info@altabioscience.com.
References
[1] Gao, Hai, et al. “Structural and Optical Properties of Zinc‐Cadmium Sulfide Nanocrystals.” Angewandte Chemie International Edition, vol. 62, no. 48, 2023, Article e202308251. Wiley Online Library, https://doi.org/10.1002/anie.202308251.
[2] PepTherDia – Home, accessed online 15/10/3034
[3] Davies, John S. “An Introduction to Cyclic Peptides.” Cyclic Peptides, edited by Jesko Koehnke, Royal Society of Chemistry, 2022, pp. 1-25. https://doi.org/10.1039/9781788010153-00001
[4] Dougherty PG, Sahni A, Pei D. Understanding Cell Penetration of Cyclic Peptides. Chem Rev. 2019 Sep 11;119(17):10241-10287. doi: 10.1021/acs.chemrev.9b00008. Epub 2019 May 14. PMID: 31083977; PMCID: PMC6739158.
[5] Natia Tsomaia, Peptide therapeutics: Targeting the undruggable space, European Journal of Medicinal Chemistry, Volume 94, 2015, Pages 459-470, ISSN 0223-5234, https://doi.org/10.1016/j.ejmech.2015.01.014.
[6] Hosam Alden Baksamawi, Alessio Alexiadis, Daniele Vigolo and Alexander Brill, “Platelet accumulation in an endothelium-coated elastic vein valve model of deep vein thrombosis is mediated by GPIbα—VWF interaction.” Frontiers in Cardiovascular Medicine, vol. 10, 2023, Article 1167884, https://doi.org/10.3389/fcvm.2023.1167884.
[7] Paul A. McEwan, Robert K. Andrews, Jonas Emsley, Glycoprotein Ibα inhibitor complex structure reveals a combined steric and allosteric mechanism of von Willebrand factor antagonism, Blood. 2009;114(23): 4883–4885. https://doi.org/10.1182/blood-2009-05-224170
[8] P.A Mcewan, Robert K Andrews, Jonas Emsley, Glycoprotein Ibalpha Inhibitor Complex Crystal Structure at High Resolution., Blood, Volume 114, Issue 22, 2009, Page 774, ISSN 0006-4971, https://doi.org/10.1182/blood.V114.22.774.774.


